В чем отличия олигофрении и деменции? Диагностика и лечение деменции в Москве
Содержание↓[показать]
Интеллектуальные способности являются самой сложной функцией мозга, благодаря которой мы можем воспринимать информацию, анализировать ее, обучаться новым навыкам, говорить, запоминать и т.п. Но существуют определенные заболевания, которые сопровождаются недостатком интеллекта, в частности это олигофрения и деменция. Эти болезни имеют схожие клинические признаки, поэтому часто их трудно дифференцировать друг от друга, хотя это очень важно, так как принципы лечения патологии и тактики ведения пациентов имеют существенные различия.
Понятия олигофрения и деменция
Олигофрения – это врожденное слабоумие, которое является необратимым и проявляется в раннем возрасте (до трех лет). Точные причины болезни не известны, но выделяется ряд провоцирующих факторов:
- гормональная недостаточность беременных женщин, в частности дефицит йода и гипофункция щитовидной железы;
- расстройства обмена веществ;
- хромосомные аномалии;
- инфекционные болезни матери во время вынашивания плода;
- гипоксия плода и т.
 п.
п.
 п.
п.Деменция – это приобретенное слабоумие, формирующееся в результате органического поражения мозговой ткани, процесс может быть обратимым или необратимым, патология характеризуется прогрессированием и постепенной утратой памяти, речи, мышления, навыков, изменением личности. Причиной болезни могут быть инфекции, травмы, сосудистые расстройства, дегенеративные процессы, наследственная предрасположенность и т.п. Чаще болезнь диагностируется в пожилом возрасте, в редких случаях регистрируется у детей.
Чем отличается олигофрения от деменции
Основные отличия деменции от олигофрении:
- олигофрения является врожденной патологией, поэтому развивается в раннем возрасте – до 3 лет, деменция чаще всего проявляется в пожилом возрасте, в редких случаях заболевание регистрируют в младшем возрасте после 2- лет;
- деменция развивается в результате повреждения нормально сформированного мозга и личности, поэтому такие дети в младшем возрасте не отличаются от здоровых малышей и нормально развиваются, олигофрения характеризуется нарушением умственного и физического развития ребенка с самого рождения;
- деменция — это прогрессирующее заболевание, для которого характерно постепенное угасание интеллектуальной функции, олигофрения является не прогрессирующей патологией;
- олигофрения характеризуется тотальностью поражения, то есть у ребенка нарушены все стороны психической деятельности, при деменции страдают определенные участки головного мозга, поэтому серьезные расстройства в одной области сочетаются с сохранением определенных функций;
- при олигофрении зачастую регистрируют положительную динамику, улучшение состояния, при деменции даже при наличии рациональной терапии часто происходит нарастание слабоумия;
- в основе деменции лежат процессы распада и снижения когнитивных функций, при олигофрении – общее недоразвитие интеллекта и познавательной деятельности.

Олигофрения и деменция: возможные результаты лечения
При постановке диагноза олигофрении или деменции не стоит отчаиваться и паниковать, необходимо как можно раньше принять наличие заболевания, чтобы предпринять все меры по предотвращению развития негативных последствий и улучшению качества жизни пациентов и их близких. Уход за больными с интеллектуальными нарушениями отнимает много времени и сил, поэтому к нему стоит морально подготовиться.
При появлении первых тревожных признаков нарушения когнитивных функций важно своевременно обратиться к врачу, так как рациональная программа терапии и тактика ведения пациентов наиболее эффективны на ранних стадиях заболеваний.
Благодаря многолетнему опыту неврологи Юсуповской больницы могут распознать наличие заболеваний на ранних этапах, для подтверждения диагноза они используют различные диагностические процедуры. При подтверждении диагноза олигофрении неврологи совместно с психотерапевтами разрабатывают индивидуальную программу лечения, которая помогает социально адаптировать пациентов, улучшая качество их жизни. При регистрации деменции также формируется индивидуальный план терапии, который зависит от причин заболевания, проявившихся симптомов и общего состояния больных. Записаться на прием к врачу можно по телефону.
При регистрации деменции также формируется индивидуальный план терапии, который зависит от причин заболевания, проявившихся симптомов и общего состояния больных. Записаться на прием к врачу можно по телефону.
Фенилкетонурия
Фенилкетонурия – наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных. В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез фенилкетонурии
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
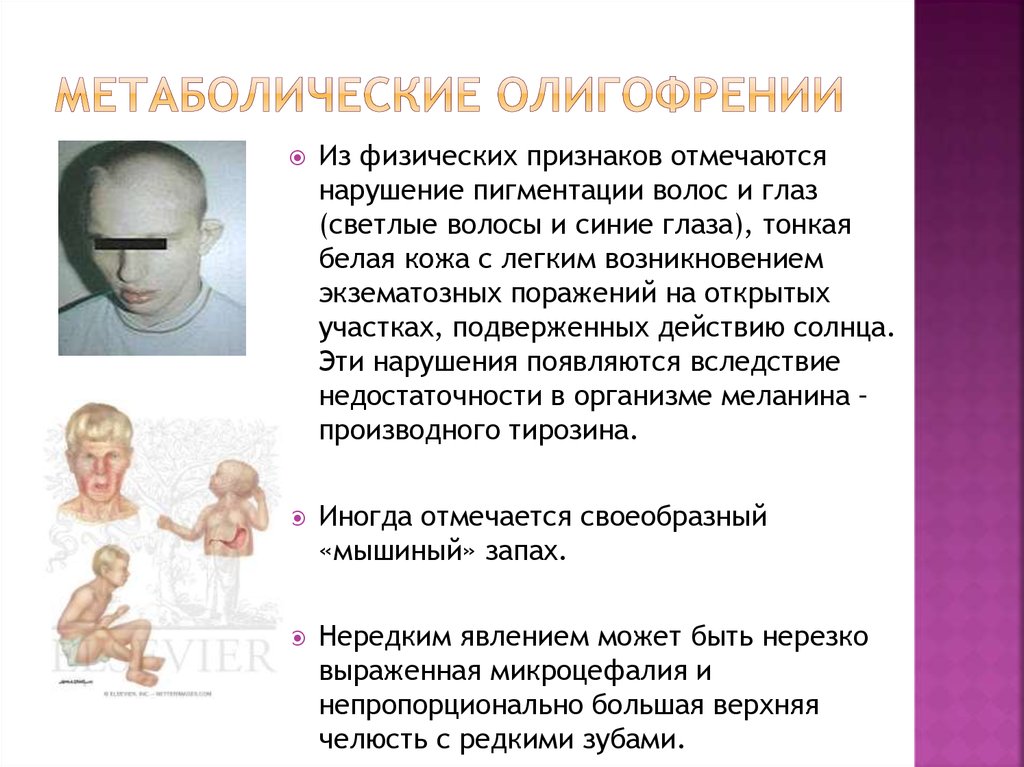
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика фенилкетонурии
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.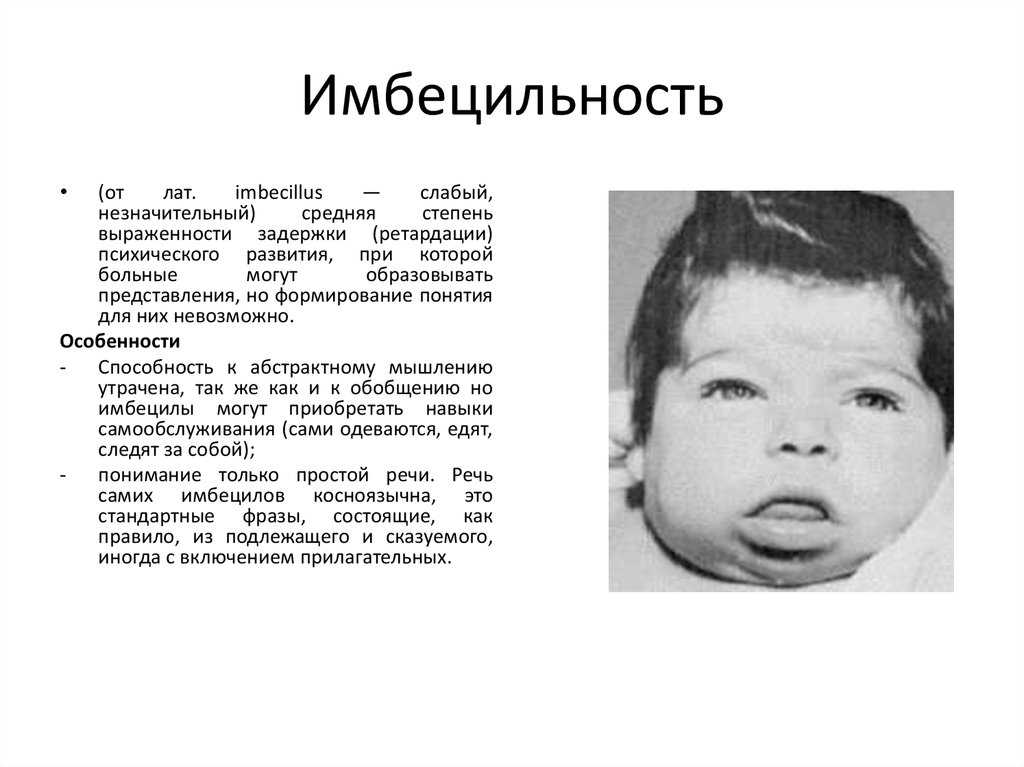
Прогноз и профилактика фенилкетонурии
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА ОЛИГОФРЕНИИ | JAMA Pediatrics
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА ОЛИГОФРЕНИИ | ДЖАМА Педиатрия | Сеть ДЖАМА [Перейти к навигации]Эта проблема
- Скачать PDF
- Полный текст
Поделиться
Твиттер Фейсбук Эл. адрес LinkedIn
- Процитировать это
- Разрешения
Артикул
Февраль 1952 г.
РОБЕРТ ГИБСОН, M.D., D.P.M.
Принадлежность авторов
ST. ЭНДРЮС, ШОТЛАНДИЯ
AMA Am J Dis Child. 1952;83(2):151-153. дои: 10.1001/archpedi.1952.02040060017001
Полный текст
Абстрактный
ПРИ диагностике умственной отсталости, зависящей не только от медицинских и психологических критериев, но и от социальных стандартов, следует отличать дефект от значительного числа других состояний. Их удобно классифицировать под заголовками интеллектуальных дефектов менее угрожающего характера, специфических дефектов зрительного, слухового или речевого характера и поведенческих дефектов, при которых аберрантное поведение, вероятно, можно спутать с психическим дефектом. Следующее расположение может служить для обозначения степени этих условий:
I.
Delayed development of speech
Congenital motor aphasia
Behavior defects
Delinquency
Maladjustment
Adolescent instability
Полный текст
Добавить или изменить учреждение
- Академическая медицина
- Кислотно-основное, электролиты, жидкости
- Аллергия и клиническая иммунология
- Анестезиология
- Антикоагулянты
- Искусство и изображения в психиатрии
- Кровотечение и переливание
- Кардиология
- Уход за тяжелобольным пациентом
- Проблемы клинической электрокардиографии
- Клиническая задача
- Поддержка принятия клинических решений
- Клинические последствия базовой нейронауки
- Клиническая фармация и фармакология
- Дополнительная и альтернативная медицина
- Заявления о консенсусе
- Коронавирус (COVID-19)
- Медицина интенсивной терапии
- Культурная компетентность
- Стоматология
- Дерматология
- Диабет и эндокринология
- Интерпретация диагностических тестов
- Разнообразие, равенство и инклюзивность
- Разработка лекарств
- Электронные медицинские карты
- Скорая помощь
- Конец жизни
- Гигиена окружающей среды
- Этика
- Пластическая хирургия лица
- Гастроэнтерология и гепатология
- Генетика и геномика
- Геномика и точное здоровье
- Гериатрия
- Глобальное здравоохранение
- Руководство по статистике и методам
- Рекомендации
- Заболевания волос
- Модели медицинского обслуживания
- Экономика здравоохранения, страхование, оплата
- Качество медицинской помощи
- Реформа здравоохранения
- Медицинская безопасность
- Медицинские работники
- Различия в состоянии здоровья
- Несправедливость в отношении здоровья
- Информатика здравоохранения
- Политика здравоохранения
- Гематология
- История медицины
- Гуманитарные науки
- Гипертония
- Изображения в неврологии
- Наука внедрения
- Инфекционные болезни
- Инновации в оказании медицинской помощи
- JAMA Инфографика
- Право и медицина
- Ведущее изменение
- Меньше значит больше
- ЛГБТК-медицина
- Образ жизни
- Медицинский код
- Медицинские приборы и оборудование
- Медицинское образование
- Медицинское образование и обучение
- Медицинские журналы и публикации
- Меланома
- Мобильное здравоохранение и телемедицина
- Нарративная медицина
- Нефрология
- Неврология
- Неврология и психиатрия
- Примечательные примечания
- Сестринское дело
- Питание
- Питание, Ожирение, Упражнения
- Ожирение
- Акушерство и гинекология
- Гигиена труда
- Онкология
- Офтальмологические изображения
- Офтальмология
- Ортопедия
- Отоларингология
- Лекарство от боли
- Патология и лабораторная медицина
- Уход за пациентами
- Информация для пациентов
- Педиатрия
- Повышение производительности
- Показатели эффективности
- Периоперационный уход и консультации
- Фармакоэкономика
- Фармакоэпидемиология
- Фармакогенетика
- Фармация и клиническая фармакология
- Физическая медицина и реабилитация
- Физиотерапия
- Руководство врача
- Поэзия
- Здоровье населения
- Профилактическая медицина
- Профессиональное благополучие
- Профессионализм
- Психиатрия и поведенческое здоровье
- Общественное здравоохранение
- Легочная медицина
- Радиология
- Регулирующие органы
- Исследования, методы, статистика
- Реанимация
- Ревматология
- Управление рисками
- Научные открытия и будущее медицины
- Совместное принятие решений и общение
- Медицина сна
- Спортивная медицина
- Трансплантация стволовых клеток
- Наркомания и наркология
- Хирургия
- Хирургические инновации
- Хирургический жемчуг
- Обучаемый момент
- Технологии и финансы
- Искусство JAMA
- Искусство и медицина
- Рациональное клиническое обследование
- Табак и электронные сигареты
- Токсикология
- Травмы и травмы
- Приверженность лечению
- УЗИ
- Урология
- Руководство пользователя по медицинской литературе
- Вакцинация
- Венозная тромбоэмболия
- Здоровье ветеранов
- Насилие
- Женское здоровье
- Рабочий процесс и процесс
- Уход за ранами, инфекция, лечение
Сохранить настройки
Политика конфиденциальности | Условия использования
Вхождение — 203600 — АЛОПЕЦИЯ-ЭПИЛЕПСИЯ-ОЛИГОФРЕНИЯ СИНДРОМ МОЙНАХАНА
Ищете этот ген или этот фенотип в других ресурсах?» qtip_text=»Выберите связанный ресурс из выпадающего меню и нажмите на целевую ссылку на непосредственно относящуюся к делу информацию. «>
«>
МКБ+
203600
АЛОПЕЦИЯ-ЭПИЛЕПСИЯ-ОЛИГОФРЕНИЯ СИНДРОМ МОЙНАХАНА
Альтернативные названия; символы
СИНДРОМ АЛОПЕЦИИ МОЙНАХАНА
Клинический обзор
▼ Ищете дополнительные ссылки?» qtip_text=»Нажмите значок «ссылка плюс»
 «> ТЕКСТ
«> ТЕКСТ В семье, о которой сообщил Moynahan (1962), пострадало 2 брата. Алопеция заключалась в задержке роста волос. Отец мальчиков был лысым до 2 лет, а тетя по материнской линии — до 4 лет. Mosavy (1975) наблюдал 4 больных сибсов, а Pfeiffer и Volklein (1982) сообщали о больных брате и сестре. Вессель и др. (1987) сообщили о 3 сибсах с алопецией, судорогами и умственной отсталостью. В семье выходцев из Бангладеш van Haeringen et al. (1990) описал отца и 3 детей (из 7) с микроцефалией, редкими волосами, умственной отсталостью от легкой до умеренной, а у 2 пострадавших мальчиков — генерализованными судорогами. Больной отец был родственником своей жены, так что это может быть примером псевдодоминирования рецессивного расстройства. Это заболевание трудно отличить от описанного в статье 203650.
▼
ССЫЛКИМосави, С.
Х. Универсальная алопеция и микроцефалия у 4 братьев и сестер. С. Афр. Мед. J. 49: 172 только, 1975.
[PubMed: 1124456, ссылки по теме]Мойнахан, Э.Дж. Семейная врожденная алопеция, эпилепсия, умственная отсталость с необычными электроэнцефалограммами.
проц. Рой. соц. Мед. 55: 411-412, 1962.
[PubMed: 14476762, ссылки по теме]Пфайффер, Р. А., Волклейн, Дж. Врожденная универсальная алопеция, умственная отсталость и микроцефалия у двух сибсов.
Дж. Мед. Жене. 19: 388-389, 1982.
[PubMed: 7143396, ссылки по теме]
[Полный текст]ван Херинген А., Херст Дж.
А., Сэвидж Р., Барайтсер М. Семейный синдром микроцефалии, редких волос, умственной отсталости и судорог. Дж. Мед. Жене. 27: 127-129, 1990.
[PubMed: 2319580, ссылки по теме]
[Полный текст]Вессель, Х.
Б., Бармада, М. А., Хашида, Ю. Врожденная алопеция, судороги и задержка психомоторного развития у трех братьев и сестер. Педиат. Нейрол. 3: 101-107, 1987.
[PubMed: 3334010, ссылки по теме]
[Полный текст]
 Х. Универсальная алопеция и микроцефалия у 4 братьев и сестер. С. Афр. Мед. J. 49: 172 только, 1975.
[PubMed: 1124456, ссылки по теме]
Х. Универсальная алопеция и микроцефалия у 4 братьев и сестер. С. Афр. Мед. J. 49: 172 только, 1975.
[PubMed: 1124456, ссылки по теме] проц. Рой. соц. Мед. 55: 411-412, 1962.
[PubMed: 14476762, ссылки по теме]
проц. Рой. соц. Мед. 55: 411-412, 1962.
[PubMed: 14476762, ссылки по теме] Дж. Мед. Жене. 19: 388-389, 1982.
[PubMed: 7143396, ссылки по теме]
[Полный текст]
Дж. Мед. Жене. 19: 388-389, 1982.
[PubMed: 7143396, ссылки по теме]
[Полный текст] А., Сэвидж Р., Барайтсер М. Семейный синдром микроцефалии, редких волос, умственной отсталости и судорог. Дж. Мед. Жене. 27: 127-129, 1990.
[PubMed: 2319580, ссылки по теме]
[Полный текст]
А., Сэвидж Р., Барайтсер М. Семейный синдром микроцефалии, редких волос, умственной отсталости и судорог. Дж. Мед. Жене. 27: 127-129, 1990.
[PubMed: 2319580, ссылки по теме]
[Полный текст] Б., Бармада, М. А., Хашида, Ю. Врожденная алопеция, судороги и задержка психомоторного развития у трех братьев и сестер. Педиат. Нейрол. 3: 101-107, 1987.
[PubMed: 3334010, ссылки по теме]
[Полный текст]
Б., Бармада, М. А., Хашида, Ю. Врожденная алопеция, судороги и задержка психомоторного развития у трех братьев и сестер. Педиат. Нейрол. 3: 101-107, 1987.
[PubMed: 3334010, ссылки по теме]
[Полный текст]Дата создания:
Victor A. McKusick : 02. 06.1986
06.1986
История редактирования:
terry : 20.05.1999
alopez : 10.06.1992 9:0443 supermimol6 : 29.02.1992
supermim : 20.03.1990
supermim : 07.03.1990
supermim : 28.02.1990
ddp : 26.10.1989
203600
АЛОПЕЦИЯ-ЭПИЛЕПСИЯ-ОЛИГОФРЕНИЯ СИНДРОМ МОЙНАХАНА
Альтернативные названия; символы
СИНДРОМ АЛОПЕЦИИ МОЙНАХАНА
SNOMEDCT: 788417006; ОРФА: 2574;
ТЕКСТ В семье, о которой сообщил Moynahan (1962), пострадало 2 брата. Алопеция заключалась в задержке роста волос. Отец мальчиков был лысым до 2 лет, а тетя по материнской линии — до 4 лет. Мосави (19 лет).75) наблюдали 4 пострадавших братьев и сестер, а Pfeiffer и Volklein (1982) сообщили о больных брате и сестре. Вессель и др. (1987) сообщили о 3 сибсах с алопецией, судорогами и умственной отсталостью. В семье выходцев из Бангладеш van Haeringen et al. (1990) описали отца и 3 детей (из 7) с микроцефалией, редкими волосами, умственной отсталостью от легкой до умеренной, а у 2 пострадавших мальчиков — генерализованными судорогами. Больной отец был родственником своей жены, так что это может быть примером псевдодоминирования рецессивного расстройства. Это расстройство трудно отличить от расстройства, описанного в статье 203650.
Алопеция заключалась в задержке роста волос. Отец мальчиков был лысым до 2 лет, а тетя по материнской линии — до 4 лет. Мосави (19 лет).75) наблюдали 4 пострадавших братьев и сестер, а Pfeiffer и Volklein (1982) сообщили о больных брате и сестре. Вессель и др. (1987) сообщили о 3 сибсах с алопецией, судорогами и умственной отсталостью. В семье выходцев из Бангладеш van Haeringen et al. (1990) описали отца и 3 детей (из 7) с микроцефалией, редкими волосами, умственной отсталостью от легкой до умеренной, а у 2 пострадавших мальчиков — генерализованными судорогами. Больной отец был родственником своей жены, так что это может быть примером псевдодоминирования рецессивного расстройства. Это расстройство трудно отличить от расстройства, описанного в статье 203650.
Мосави, С.
Х. Универсальная алопеция и микроцефалия у 4 братьев и сестер. С. Афр. Мед. J. 49: 172 только, 1975.
[В паблике: 1124456]Мойнахан, Э.Дж. Семейная врожденная алопеция, эпилепсия, умственная отсталость с необычными электроэнцефалограммами.
проц. Рой. соц. Мед. 55: 411-412, 1962.
[В паблике: 14476762]Пфайффер, Р. А., Волклейн, Дж. Врожденная универсальная алопеция, умственная отсталость и микроцефалия у двух сибсов. Дж. Мед. Жене. 19: 388-389, 1982. [В паблике: 7143396] [Полный текст: https://doi.
org/10.1136/jmg.19.5.388]ван Херинген А., Херст Дж. А., Сэвидж Р., Барайтсер М. Семейный синдром микроцефалии, редких волос, умственной отсталости и судорог. Дж. Мед. Жене. 27: 127-129, 1990. [В паблике: 2319580] [Полный текст: https://doi.
org/10.1136/jmg.27.2.127]Вессель, Х. Б., Бармада, М. А., Хашида, Ю. Врожденная алопеция, судороги и задержка психомоторного развития у трех братьев и сестер. Педиат. Нейрол. 3: 101-107, 1987. [Пубмед: 3334010] [Полный текст: https://doi.
 Х. Универсальная алопеция и микроцефалия у 4 братьев и сестер. С. Афр. Мед. J. 49: 172 только, 1975.
[В паблике: 1124456]
Х. Универсальная алопеция и микроцефалия у 4 братьев и сестер. С. Афр. Мед. J. 49: 172 только, 1975.
[В паблике: 1124456] проц. Рой. соц. Мед. 55: 411-412, 1962.
[В паблике: 14476762]
проц. Рой. соц. Мед. 55: 411-412, 1962.
[В паблике: 14476762] org/10.1136/jmg.19.5.388]
org/10.1136/jmg.19.5.388] org/10.1136/jmg.27.2.127]
org/10.1136/jmg.27.2.127]